



An Introduction to Amplicon Sequencing of 16S/18S/ITS Regions

Posted by kikoetgarcia
from the Agriculture category at
28 Jun 2023 09:39:19 am.
Share this page: 












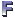






Amplicon sequencing is a targeted approach that focuses on specific regions of DNA or RNA within a given sample. It involves amplifying and sequencing a particular gene or genomic region of interest. In the field of microbial ecology, the 16S rRNA gene is commonly targeted for bacteria and archaea. This gene contains conserved regions that allow for the design of universal primers, enabling amplification and sequencing of a wide range of microbial taxa. By sequencing the 16S rRNA gene, researchers can determine the taxonomic composition of the microbial community present in the sample. While amplicon sequencing provides information about the diversity and relative abundance of different taxa, it has limitations in providing functional information.
How does Amplicon Sequencing differ from Metagenomics?
Metagenomic sequencing, on the other hand, involves sequencing the entire genetic material (DNA or RNA) present in a sample, without specifically targeting regions of interest. It provides a comprehensive snapshot of the entire microbial community, including both known and unknown microorganisms. Metagenomic sequencing not only identifies the taxonomic composition of the community but also reveals the functional potential of the microorganisms present. By analyzing the entire genetic material, researchers can study the complete microbial genome and identify specific genes and pathways involved in various functions such as metabolism, antibiotic resistance, and virulence. Metagenomic sequencing offers higher resolution in taxonomic identification and functional potential compared to amplicon sequencing but can be computationally and financially demanding.
In summary, amplicon sequencing provides information about the taxonomic composition and relative abundance of specific microbial taxa within a community, while metagenomic sequencing provides a more comprehensive view of the entire microbial community, including taxonomic information and functional potential. The choice between amplicon sequencing and metagenomic sequencing depends on the research objectives, available resources, and the level of detail required for the analysis.
Sanger sequencing vs. NGS-based targeted sequencing vs. long-read sequencing
Amplicon sequencing, which encompasses Sanger sequencing and next-generation sequencing (NGS) approaches, has become a powerful tool for studying microbial communities and targeted genetic analysis. Understanding the strengths and limitations of each technique is crucial for designing effective experimental strategies and accurately interpreting results.
Sanger sequencing, based on chain termination principles, provides reliable and highly accurate sequencing data, making it an ideal choice for confirming and validating results obtained from larger-scale amplicon sequencing projects. However, due to its low throughput and time-consuming nature, Sanger sequencing is less suitable for high-throughput studies involving a large number of DNA fragments.
Next-generation sequencing platforms, such as Illumina sequencing, offer increased throughput and reduced per-sample costs by employing strategies like amplicon sequencing or hybrid capture to selectively enrich the regions of interest. Multiplexing samples, where multiple samples are pooled and sequenced together, significantly enhances throughput and facilitates comprehensive analysis of complex genetic variations and diverse microbial communities.
Furthermore, the emergence of long-read sequencing technologies, such as PacBio sequencing and Oxford Nanopore sequencing, should be considered in the context of amplicon sequencing. Long-read sequencing provides advantages such as significantly longer sequencing reads, enabling applications like de novo genome assembly, haplotype phasing, and characterization of complex genomic rearrangements. However, it is important to note that long-read sequencing technologies generally have higher error rates and limitations in terms of throughput and cost per base.
Each method has its own strengths and considerations, requiring careful assessment of research objectives, sample size, desired coverage, and available resources.
Bacterial 16S rRNA
Bacterial ribosomal RNA (rRNA) consists of three types: 5S rRNA (120 bp), 16S rRNA (approximately 1540 bp), and 23S rRNA (approximately 2900 bp). The 16S rRNA is commonly found in prokaryotic cells and accounts for more than 80% of total bacterial RNA. It has a high copy number, easy accessibility to templates, high functional homology, and moderate genetic information.
The 16S rRNA coding gene sequence contains nine conserved regions and nine highly variable regions. Among them, the V3-V4 region offers good specificity, complete database information, and is the optimal choice for bacterial diversity analysis annotation.
Eukaryotic 18S rRNA
The 18S rRNA gene is a DNA sequence that encodes a small subunit of eukaryotic ribosomes. It consists of both conserved and variable regions (V1-V9, excluding V6 region). Among these regions, the V4 region is widely used, providing the most comprehensive database information and optimal classification for analyzing the 18S rRNA gene.
Archaeal 16S rRNA
Archaebacteria, also known as archaea, are a unique class of bacteria that exhibit characteristics of both prokaryotes and eukaryotes. For the Illumina 2x250 bp sequencing platform, the primer set 519F/915R is highly suitable for archaeal 16S rRNA analysis.
Fungal ITS sequences
Internally Transcribed Spacer (ITS) sequences are located between fungal 18S, 5.8S, and 28S rRNA genes, specifically ITS1 and ITS2. These ITS sequence fragments are relatively small (350 bp for ITS1 and 400 bp for ITS2), facilitating analysis, and widely used for phylogenetic analysis of different fungal species.
Diversity analysis of specific functional microorganisms
Functional microorganisms are of great interest due to their importance in various ecological processes, such as nitrifying bacteria, denitrifying bacteria, ammonia-oxidizing bacteria, sulfate-reducing bacteria, and nitrogen-fixing bacteria.
Although each functional microorganism may be taxonomically diverse, they possess similar genes that enable them to perform specific functions. These genes, responsible for specific functions in functional bacteria, are known as functional genes. Examples of functional genes include nxrA, nirS/nirK, amoA, dsrB, and nifH.
2 Comments
Comments

nụklasts
fallguys
, you can play afun race with your friends or family after a long day at work. This game is funfor everyone because it has cute graphics and is fun to play.<o:p></o:p>